Boronic acid

The general structure of a boronic acid, where R is a substituent.
A boronic acid is a compound related to boric acid in which one of the three hydroxyl groups is replaced by an alkyl or aryl group.[1] As a compound containing a carbon–boron bond, members of this class thus belong to the larger class of organoboranes. Boronic acids act as Lewis acids. Their unique feature is that they are capable of forming reversible covalent complexes with sugars, amino acids, hydroxamic acids, etc. (molecules with vicinal, (1,2) or occasionally (1,3) substituted Lewis base donors (alcohol, amine, carboxylate)). The pKa of a boronic acid is ~9, but they can form tetrahedral boronate complexes with pKa ~7. They are occasionally used in the area of molecular recognition to bind to saccharides for fluorescent detection or selective transport of saccharides across membranes.
Boronic acids are used extensively in organic chemistry as chemical building blocks and intermediates predominantly in the Suzuki coupling. A key concept in its chemistry is transmetallation of its organic residue to a transition metal.
The compound bortezomib with a boronic acid group is a drug used in chemotherapy. The boron atom in this molecule is a key substructure because through it certain proteasomes are blocked that would otherwise degrade proteins. Boronic acids are known to bind to active site serines and are part of inhibitors for porcine pancreatic lipase,[2]subtilisin[3] and the protease Kex2.[4] Furthermore, boronic acid derivatives constitute a class of inhibitors for human acyl-protein thioesterase 1 and 2, which are cancer drug targets within the Ras cycle.[5]
The boronic acid functional group is reputed to have low inherent toxicity. This is one of the reasons for the popularity of the Suzuki coupling in the development and synthesis of pharmaceutical agents. However, a significant fraction of commonly used boronic acids and their derivatives were recently found to gives a positive Ames test and act as chemical mutagens. The mechanism of mutagenicity is thought to involve the generation of organic radicals via oxidation of the boronic acid by atmospheric oxygen.[6]
Contents
1 Structure and synthesis
1.1 Synthesis
2 Boronic esters (also named boronate esters)
3 Organic chemistry applications
3.1 Suzuki coupling reaction
3.2 Chan–Lam coupling
3.3 Liebeskind–Srogl coupling
3.4 Conjugate addition
3.5 Oxidation
3.6 Homologation
3.7 Electrophilic allyl shifts
3.8 Hydrolysis
3.9 C–H coupling reactions
3.10 Protonolysis
4 Supramolecular chemistry
4.1 Saccharide recognition
5 Notes
6 References
7 External links
Structure and synthesis
In 1860, Edward Frankland was the first to report the preparation and isolation of a boronic acid. Ethylboronic acid was synthesized by a two-stage process. First, diethylzinc and triethyl borate reacted to produce triethylborane. This compound then oxidized in air to form ethylboronic acid.[7][8][9] Several synthetic routes are now in common use, and many air-stable boronic acids are commercially available.
Boronic acids typically have high melting points. They are prone to forming anhydrides by loss of water molecules, typically to give cyclic trimers.
| Boronic acid | R | Structure | Molar mass | CAS number | Melting point °C |
|---|---|---|---|---|---|
| Phenylboronic acid | Phenyl |  | 121.93 | 98-80-6 | 216–219 |
| 2-Thienylboronic acid | Thiophen |  | 127.96 | 6165-68-0 | 138–140 |
| Methylboronic acid | Methyl |  | 59.86 | 13061-96-6 | 91–94 |
cis-Propenylboronic acid | propene | 85.90 | 7547-96-8 | 65–70 | |
trans-Propenylboronic acid | propene | 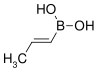 | 85.90 | 7547-97-9 | 123–127 |
Synthesis
Boronic acids can be obtained via several methods. The most common way is reaction of organometallic compounds based on lithium or magnesium (Grignards) with borate esters.[10][11][12][13] For example, phenylboronic acid is produced from phenylmagnesium bromide and trimethyl borate followed by hydrolysis[14]
- PhMgBr + B(OMe)3 → PhB(OMe)2 + MeOMgBr
- PhB(OMe)2 + H2O → PhB(OH)2 + MeOH
Another method is reaction of an arylsilane (RSiR3) with boron tribromide (BBr3) in a transmetallation to RBBr2 followed by acidic hydrolysis.
A third method is by palladium catalysed reaction of aryl halides and triflates with diboronyl esters in a coupling reaction. An alternative to esters in this method is the use of diboronic acid or tetrahydroxydiboron ([B(OH2)]2).[15][16]
Boronic esters (also named boronate esters)
Boronic esters are esters formed between a boronic acid and an alcohol.
| Compound | General formula | General structure |
|---|---|---|
| Boronic acid | RB(OH)2 |  |
| Boronic ester | RB(OR)2 |  |
The compounds can be obtained from borate esters[17] by condensation with alcohols and diols. Phenylboronic acid can be selfcondensed to the cyclic trimer called triphenyl anhydride or triphenylboroxin.[18]
| Boronic ester | Diol | Structural formula | Molar mass | CAS number | Boiling point (°C) |
|---|---|---|---|---|---|
| Allylboronic acid pinacol ester | pinacol |  | 168.04 | 72824-04-5 | 50–53 (5 mmHg) |
| Phenyl boronic acid trimethylene glycol ester | trimethylene glycol |  | 161.99 | 4406-77-3 | 106 (2 mm Hg) |
| Diisopropoxymethylborane | isopropanol | 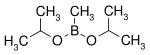 | 144.02 | 86595-27-9 | 105 -107 |
Compounds with 5-membered cyclic structures containing the C–O–B–O–C linkage are called dioxaborolanes and those with 6-membered rings dioxaborinanes.
Organic chemistry applications
Suzuki coupling reaction
Boronic acids are used in organic chemistry in the Suzuki reaction. In this reaction the boron atom exchanges its aryl group with an alkoxy group from palladium.
R1−BY2+R2−X→BasePdcatalystR1−R2{displaystyle {begin{matrix}{}\{ce {{R1-BY2}+R2-X->[{underset {text{catalyst}}{text{Pd}}}][{text{Base}}]R1-R2}}\{}end{matrix}}}
(1)
![{displaystyle {begin{matrix}{}\{ce {{R1-BY2}+R2-X->[{underset {text{catalyst}}{text{Pd}}}][{text{Base}}]R1-R2}}\{}end{matrix}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c35df36f38fec8abbc8e9d1d9f04e9b2687ae245 "The Suzuki reaction")
Chan–Lam coupling
In the Chan–Lam coupling the alkyl, alkenyl or aryl boronic acid reacts with a N–H or O–H containing compound with Cu(II) such as copper(II) acetate and oxygen and a base such as pyridine[19][20] forming a new carbon–nitrogen bond or carbon–oxygen bond for example in this reaction of 2-pyridone with trans-1-hexenylboronic acid:

The reaction mechanism sequence is deprotonation of the amine, coordination of the amine to the copper(II), transmetallation (transferring the alkyl boron group to copper and the copper acetate group to boron), oxidation of Cu(II) to Cu(III) by oxygen and finally reductive elimination of Cu(III) to Cu(I) with formation of the product. Direct reductive elimination of Cu(II) to Cu(0) also takes place but is very slow. In catalytic systems oxygen also regenerates the Cu(II) catalyst.
Liebeskind–Srogl coupling
In the Liebeskind–Srogl coupling a thiol ester is coupled with a boronic acid to produce a ketone.
Conjugate addition
The boronic acid organic residue is a nucleophile in conjugate addition also in conjunction with a metal. In one study the pinacol ester of allylboronic acid is reacted with dibenzylidene acetone in such a conjugate addition:[21]

- The catalyst system in this reaction is tris(dibenzylideneacetone)dipalladium(0) / tricyclohexylphosphine.
Another conjugate addition is that of gramine with phenylboronic acid catalyzed by cyclooctadiene rhodium chloride dimer:[22]

Oxidation
Boronic esters are oxidized to the corresponding alcohols with base and hydrogen peroxide (for an example see: carbenoid)
Homologation
- In boronic ester homologization an alkyl group shifts from boron in a boronate to carbon:[23]

Boronic ester homologization

Homologization application


In this reaction dichloromethyllithium converts the boronic ester into a boronate. A Lewis acid then induces a rearrangement of the alkyl group with displacement of the chlorine group. Finally an organometallic reagent such as a Grignard reagent displaces the second chlorine atom effectively leading to insertion of an RCH2 group into the C-B bond. Another reaction featuring a boronate alkyl migration is the Petasis reaction.
Electrophilic allyl shifts
Allyl boronic esters engage in electrophilic allyl shifts very much like silicon pendant in the Sakurai reaction. In one study a diallylation reagent combines both[24][note 1]:

Hydrolysis
Hydrolysis of boronic esters back to the boronic acid and the alcohol can be accomplished in certain systems with thionyl chloride and pyridine.[25]
Aryl boronic acids or esters may be hydrolyzed to the corresponding phenols by reaction with hydroxylamine at room temperature.[26]
C–H coupling reactions
The diboron compound bis(pinacolato)diboron[27] reacts with aromatic heterocycles[28] or simple arenes[29] to an arylboronate ester with iridium catalyst [IrCl(COD)]2 (a modification of Crabtree's catalyst) and base 4,4′-di-tert-butyl-2,2′-bipyridine in a C-H coupling reaction for example with benzene:

In one modification the arene reacts using only a stoichiometric equivalent rather than a large excess using the cheaper pinacolborane:[30]

Unlike in ordinary electrophilic aromatic substitution (EAS) where electronic effects dominate, the regioselectivity in this reaction type is solely determined by the steric bulk of the iridium complex. This is exploited in a meta-bromination of m-xylene which by standard AES would give the ortho product:[31][note 2]

Protonolysis
Protodeboronation is a chemical reaction involving the protonolysis of a boronic acid (or other organoborane compound) in which a carbon-boron bond is broken and replaced with a carbon-hydrogen bond. Protodeboronation is a well-known undesired side reaction, and frequently associated with metal-catalysed coupling reactions that utilise boronic acids (see Suzuki reaction). For a given boronic acid, the propensity to undergo protodeboronation is highly variable and dependent on various factors, such as the reaction conditions employed and the organic substituent of the boronic acid:

Supramolecular chemistry
Saccharide recognition

An example of a fluorescent complex of a diboronic acid and tartaric acid[32]
The covalent pair-wise interaction between boronic acids and hydroxy groups as found in alcohols and acids is rapid and reversible in aqueous solutions. The equilibrium established between boronic acids and the hydroxyl groups present on saccharides has been successfully employed to develop a range of sensors for saccharides.[33] One of the key advantages with this dynamic covalent strategy[34] lies in the ability of boronic acids to overcome the challenge of binding neutral species in aqueous media. If arranged correctly, the introduction of a tertiary amine within these supramolecular systems will permit binding to occur at physiological pH and allow signalling mechanisms such as photoinduced electron transfer mediated fluorescence emission to report the binding event.
Potential applications for this research include blood glucose monitoring systems to help manage diabetes mellitus. As the sensors employ an optical response, monitoring could be achieved using minimally invasive methods, one such example is the investigation of a contact lens that contains a boronic acid based sensor molecule to detect glucose levels within ocular fluids.[35]
Notes
^ In this sequence the boronic ester allyl shift is catalyzed by boron trifluoride. In the second step the hydroxyl group is activated as a leaving group by conversion to a triflate by triflic anhydride aided by 2,6-lutidine. The final product is a vinyl cyclopropane. Note: ee stands for enantiomeric excess
^ In situ second step reaction of boronate ester with copper(II) bromide
References
^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Boronic Acids". doi:10.1351/goldbook.B00714
^ Garner, C. W. (1980-06-10). "Boronic acid inhibitors of porcine pancreatic lipase". The Journal of Biological Chemistry. 255 (11): 5064–5068. ISSN 0021-9258. PMID 7372625..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output .citation q{quotes:"""""""'""'"}.mw-parser-output .citation .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .citation .cs1-lock-limited a,.mw-parser-output .citation .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .citation .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-ws-icon a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/4/4c/Wikisource-logo.svg/12px-Wikisource-logo.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-maint{display:none;color:#33aa33;margin-left:0.3em}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ Lindquist, R. N.; Terry, C. (January 1974). "Inhibition of subtilisin by boronic acids, potential analogs of tetrahedral reaction intermediates". Archives of Biochemistry and Biophysics. 160 (1): 135–144. doi:10.1016/s0003-9861(74)80018-4. ISSN 0003-9861. PMID 4364061.
^ Holyoak, Todd; Wilson, Mark A.; Fenn, Timothy D.; Kettner, Charles A.; Petsko, Gregory A.; Fuller, Robert S.; Ringe, Dagmar (2003-06-10). "2.4 A resolution crystal structure of the prototypical hormone-processing protease Kex2 in complex with an Ala-Lys-Arg boronic acid inhibitor". Biochemistry. 42 (22): 6709–6718. doi:10.1021/bi034434t. ISSN 0006-2960. PMID 12779325.
^ Zimmermann, Tobias J.; Bürger, Marco; Tashiro, Etsu; Kondoh, Yasumitsu; Martinez, Nancy E.; Görmer, Kristina; Rosin-Steiner, Sigrid; Shimizu, Takeshi; Ozaki, Shoichiro (2013-01-02). "Boron-based inhibitors of acyl protein thioesterases 1 and 2". ChemBioChem: A European Journal of Chemical Biology. 14 (1): 115–122. doi:10.1002/cbic.201200571. ISSN 1439-7633. PMID 23239555.
^ Hansen, Marvin M.; Jolly, Robert A.; Linder, Ryan J. (2015-07-29). "Boronic Acids and Derivatives—Probing the Structure–Activity Relationships for Mutagenicity". Organic Process Research & Development. 19 (11): 1507–1516. doi:10.1021/acs.oprd.5b00150. ISSN 1083-6160.
^ Frankland, E.; Duppa, B. F. (1860). "Vorläufige Notiz über Boräthyl". Justus Liebigs Ann. Chem. 115 (3): 319. doi:10.1002/jlac.18601150324.
^ Frankland, E.; Duppa, B. (1860). "On Boric Ethide". Proceedings of the Royal Society. 10: 568. doi:10.1098/rspl.1859.0112.
^ Frankland, E. (1862). "On a new series of organic compounds containing boron". J. Chem. Soc. 15: 363. doi:10.1039/JS8621500363.
^ Dennis G. Hall, ed. (2005). Boronic Acids. Wiley. ISBN 3-527-30991-8.
^ Example: Kristensen, Jesper Langgaard; Lysén, Morten; Vedsø, Per; Begtrup, Mikael (2005). "Synthesis of Ortho Substituted Arylboronic Esters by in situ Traping of Unstable Lithio Intermediates: 2-(5,5-Dimethyl-1,3,2-dioxaborinan-2-yl)benzoic acid ethyl ester". Organic Syntheses. 81: 134.; Collective Volume, 11, pp. 1015 prep= v81p0134
^ Example: Li, Wenjie; Nelson, Dorian P.; Jensen, Mark S.; Scott Hoerrner, R.; Cai, Dongwei; Larsen, Robert D. (2005). "Synthesis of 3-Pyridylboronic Acid and its Pinacol Ester. Application of 3-Pyridylboronic acid in Suzuki Coupling to Prepare 3-Pyridin-3-ylquinoline". Organic Syntheses. 81: 89.; Collective Volume, 11, p. 393
^ Charette, André B.; Lebel, Hélène (1999). "(2S,3S)-(+)-(3-Phenylcyclopropyl)methanol". Organic Syntheses. 76: 86.; Collective Volume, 10, p. 613
^ Washburn, Robert M.; Levens, Ernest; Albright, Charles F.; Billig, Franklin A. (1959). "Benzeneboronic anhydride". Organic Syntheses. 39: 3.; Collective Volume, 4, p. 68
^ Pilarski, Lukasz T.; Szabó, Kálmán J. (2011). "Palladium-Catalyzed Direct Synthesis of Organoboronic Acids". Angewandte Chemie International Edition. 50: 8230–8232. doi:10.1002/anie.201102384.
^ Molander, Gary A.; Trice, Sarah L. J.; Dreher, Spencer D. (2010). "Palladium-Catalyzed, Direct Boronic Acid Synthesis from Aryl Chlorides: A Simplified Route to Diverse Boronate Ester Derivatives". Journal of the American Chemical Society. 132 (50): 17701–17703. doi:10.1021/ja1089759. PMC 3075417.
^ Kidwell, R. L.; Murphy, M.; Darling, S. D. (1969). "Phenols: 6-Methoxy-2-Naphthol". Organic Syntheses. 49: 90.; Collective Volume, 5, p. 918
^ Washburn, Robert M.; Levens, Ernest; Albright, Charles F.; Billig, Franklin A. (1959). "Benzeneboronic anhydride". Organic Syntheses. 39: 3.; Collective Volume, 4, p. 68
^ Chan, Dominic M.T. (2003). "Copper promoted C–N and C–O bond cross-coupling with phenyl and pyridylboronates". Tetrahedron Letters. 44: 3863–3865. doi:10.1016/S0040-4039(03)00739-1.
^ Lam, Patrick Y.S (2003). "Copper-promoted/catalyzed C–N and C–O bond cross-coupling with vinylboronic acid and its utilities". Tetrahedron Letters. 44: 4927–4931. doi:10.1016/S0040-4039(03)01037-2.
^ Sieber, Joshua D. (2007). "Catalytic Conjugate Addition of Allyl Groups to Styryl-Activated Enones". Journal of the American Chemical Society. 129: 2214–2215. doi:10.1021/ja067878w.
^ Gabriela (2007). "Benzylic Substitution of Gramines with Boronic Acids and Rhodium or Iridium Catalysts †". Organic Letters. 9: 961–964. doi:10.1021/ol063042m.
^ Matteson, Donald S. (1986). "99% Chirally selective synthesis via pinanediol boronic esters: insect pheromones, diols, and an amino alcohol". Journal of the American Chemical Society. 108: 810–819. doi:10.1021/ja00264a039.
^ Peng, Feng (2007). "Simple, Stable, and Versatile Double-Allylation Reagents for the Stereoselective Preparation of Skeletally Diverse Compounds". Journal of the American Chemical Society. 129: 3070–3071. doi:10.1021/ja068985t.
^ Matteson, Donald S. (2003). "New asymmetric syntheses with boronic esters and fluoroboranes" (PDF). Pure Appl. Chem. 75 (9): 1249–1253. doi:10.1351/pac200375091249.
^ Kianmehr, Ebrahim; Yahyaee, Maryam; Tabatabai, Katayoun (2007). "A mild conversion of arylboronic acids and their pinacolyl boronate esters into phenols using hydroxylamine". Tetrahedron Letters. 48: 2713–2715. doi:10.1016/j.tetlet.2007.02.069.
^ Ishiyama, Tatsuo; Murata, Miki; Ahiko, Taka-aki; Miyaura, Norio (2000). "Bis(pinacolato)diboron". Organic Syntheses. 77: 176.; Collective Volume, 10, p. 115
^ Takagi, Jun (2002). "Iridium-catalyzed C–H coupling reaction of heteroaromatic compounds with bis(pinacolato)diboron: regioselective synthesis of heteroarylboronates". Tetrahedron Letters. 43: 5649–5651. doi:10.1016/S0040-4039(02)01135-8.
^ Ishiyama, Tatsuo (2002). "Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate". Journal of the American Chemical Society. 124: 390–391. doi:10.1021/ja0173019.
^ Ishiyama, Tatsuo (2003). "Room temperature borylation of arenes and heteroarenes using stoichiometric amounts of pinacolborane catalyzed by iridium complexes in an inert solvent". Chemical Communications (23): 2924. doi:10.1039/b311103b.
^ Murphy, Jaclyn M. (2007). "Meta Halogenation of 1,3-Disubstituted Arenes via Iridium-Catalyzed Arene Borylation". Journal of the American Chemical Society. 129: 15434–15435. doi:10.1021/ja076498n.
^ Zhao, Jianzhang; Davidson, Matthew G.; Mahon, Mary F.; Kociok-Köhn, Gabriele; James, Tony D. (2004). "An Enantioselective Fluorescent Sensor for Sugar Acids". J. Am. Chem. Soc. 126 (49): 16179–16186. doi:10.1021/ja046289s.
^ "Boronic Acids in Saccharide Recognition". 2006. doi:10.1039/9781847557612.
^ Rowan, Stuart J.; Cantrill, Stuart J.; Cousins, Graham R. L.; Sanders, Jeremy K. M.; Stoddart, J. Fraser (2002). "Dynamic Covalent Chemistry". Angewandte Chemie International Edition. 41 (6): 898–952. doi:10.1002/1521-3773(20020315)41:6<898::AID-ANIE898>3.0.CO;2-E. PMID 12491278.
^ US 6850786, Wayne Front March, "Ocular analyte sensor", issued 2005-02-01
External links
- Boronic acids database
Branches of chemistry | |
|---|---|
| |
| Physical |
|
| Organic |
|
| Inorganic |
|
| Others |
|
| |


