Cystic fibrosis transmembrane conductance regulator
| CFTR | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||
| |||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||
| Aliases | CFTR, ABC35, ABCC7, CF, CFTR/MRP, MRP7, TNR-dJ760C5.1, cystic fibrosis transmembrane conductance regulator | ||||||||||||||||||||||||
| External IDs | OMIM: 602421 MGI: 88388 HomoloGene: 55465 GeneCards: CFTR | ||||||||||||||||||||||||
| EC number | 3.6.3.49 | ||||||||||||||||||||||||
| |||||||||||||||||||||||||
| |||||||||||||||||||||||||
| |||||||||||||||||||||||||
| Orthologs | |||||||||||||||||||||||||
| Species | Human | Mouse | |||||||||||||||||||||||
| Entrez |
|
| |||||||||||||||||||||||
| Ensembl |
|
| |||||||||||||||||||||||
| UniProt |
|
| |||||||||||||||||||||||
| RefSeq (mRNA) |
|
| |||||||||||||||||||||||
| RefSeq (protein) |
|
| |||||||||||||||||||||||
| Location (UCSC) | Chr 7: 117.47 – 117.72 Mb | Chr 6: 18.17 – 18.32 Mb | |||||||||||||||||||||||
PubMed search | [3] | [4] | |||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||
| |||||||||||||||||||||||||




Cystic fibrosis transmembrane conductance regulator (CFTR) is a membrane protein and chloride channel in vertebrates that is encoded by the CFTR gene.[5][6]
The CFTR gene codes for an ABC transporter-class ion channel protein that conducts chloride[7] and thiocyanate[8] ions across epithelial cell membranes. Mutations of the CFTR gene affecting chloride ion channel function lead to dysregulation of epithelial fluid transport in the lung, pancreas and other organs, resulting in cystic fibrosis. Complications include thickened mucus in the lungs with frequent respiratory infections, and pancreatic insufficiency giving rise to malnutrition and diabetes. These conditions lead to chronic disability and reduced life expectancy. In male patients, the progressive obstruction and destruction of the developing vas deferens (spermatic cord) and epididymis appear to result from abnormal intraluminal secretions,[9] causing congenital absence of the vas deferens and male infertility.
Contents
1 Gene
1.1 Mutations
1.2 List of common mutations
2 Structure
3 Location and function
3.1 Interactions
4 Related conditions
5 Drug target
6 References
7 Further reading
8 External links
Gene

The location of the CFTR gene on chromosome 7
The gene that encodes the human CFTR protein is found on chromosome 7, on the long arm at position q31.2.[6] from base pair 116,907,253 to base pair 117,095,955. CFTR orthologs [10] occur in the jawed vertebrates.[11]
The CFTR gene has been used in animals as a nuclear DNA phylogenetic marker.[10] Large genomic sequences of this gene have been used to explore the phylogeny of the major groups of mammals,[12] and confirmed the grouping of placental orders into four major clades: Xenarthra, Afrotheria, Laurasiatheria, and Euarchonta plus Glires.
Mutations
Nearly 1000 cystic fibrosis-causing mutations have been described.[13] The most common mutation, ΔF508 results from a deletion (Δ) of three nucleotides which results in a loss of the amino acid phenylalanine (F) at the 508th position on the protein. As a result, the protein does not fold normally and is more quickly degraded. The vast majority of mutations are infrequent. The distribution and frequency of mutations varies among different populations which has implications for genetic screening and counseling.
Mutations consist of replacements, duplications, deletions or shortenings in the CFTR gene. This may result in proteins that may not function, work less effectively, are more quickly degraded, or are present in inadequate numbers.[14]
It has been hypothesized that mutations in the CFTR gene may confer a selective advantage to heterozygous individuals. Cells expressing a mutant form of the CFTR protein are resistant to invasion by the Salmonella typhi bacterium, the agent of typhoid fever, and mice carrying a single copy of mutant CFTR are resistant to diarrhea caused by cholera toxin.[15]
List of common mutations

The most common mutations among caucasians are:[16]
- ΔF508
- G542X
- G551D
- N1303K
- W1282X
Structure
The CFTR gene is approximately 189 kb in length, with 27 exons and 26 introns.[17] CFTR is a glycoprotein with 1480 amino acids. The protein consists of five domains. There are two transmembrane domains, each with six spans of alpha helices. These are each connected to a nucleotide binding domain (NBD) in the cytoplasm. The first NBD is connected to the second transmembrane domain by a regulatory "R" domain that is a unique feature of CFTR, not present in other ABC transporters. The ion channel only opens when its R-domain has been phosphorylated by PKA and ATP is bound at the NBDs.[18] The carboxyl terminal of the protein is anchored to the cytoskeleton by a PDZ-interacting domain.[19]Caveat: The crystal structure included at the top is not the full CFTR channel (the cartoon version is OK). The correct PDB accession number for the channel structure is 5UAK. The structure shown (PDB# 1XMI) shows a homopentameric assembly of mutated NBD1, the first nucleotide binding domain (NBD1) of the transporter.
Location and function
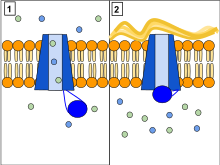
The CFTR protein is a channel protein that controls the flow of H2O and Cl− ions in and out of cells inside the lungs. When the CFTR protein is working correctly, as shown in Panel 1, ions freely flow in and out of the cells. However, when the CFTR protein is malfunctioning as in Panel 2, these ions cannot flow out of the cell due to blocked CFTR channels. This occurs in cystic fibrosis, characterized by the buildup of thick mucus in the lungs.
CFTR functions as an ATP-gated anion channel, increasing the conductance for certain anions (e.g. Cl−) to flow down their electrochemical gradient. ATP-driven conformational changes in CFTR open and close a gate to allow transmembrane flow of anions down their electrochemical gradient.[5] This in contrast to other ABC proteins, in which ATP-driven conformational changes fuel uphill substrate transport across cellular membranes. Essentially, CFTR is an ion channel that evolved as a 'broken' ABC transporter that leaks when in open conformation.
CFTRs have two transmembrane domains, whereby each have a nucleotide-binding domain attached to it. CFTRs also contain another domain called the regulatory domain, which consists of both the sections mentioned above. Other isoforms of ABC ion channels are involved in the uptake of nutrients in prokaryotes. The CFTRs have an evolutionary design to transfer the free energy of ATP hydrolysis to the uphill movement of anions across the cell membrane. The ion channels have two main conformations, one where the cargo binding site is inward facing (ATP bound), and one where it is outward facing (ATP free). ATP binds to each individual nucleotide binding domain, which results in the subsequent ATP hydrolysis, leading to the rearrangement of the transmembrane helices and transmembrane domains. This changes the accessibility of the cargo binding site to an inward facing position. This irreversible ATP binding and hydrolysis, drives the alternative exposure of the CFTR, ensuring a unidirectional transport of anions down an electrochemical gradient.[20][21]
The CFTR is found in the epithelial cells of many organs including the lung, liver, pancreas, digestive tract, and the reproductive tract. In the airways of the lung, CFTR is most highly expressed by rare specialized cells called ionocytes.[22][23] In the skin CFTR is strongly expressed in the sebaceous and eccrine sweat glands.[24] In the eccrine glands, CFTR is located on the apical membrane of the epithelial cells that make up the duct of these sweat glands.[24]
Normally, the protein moves chloride and thiocyanate[25]ions (with a negative charge) out of an epithelial cell to the covering mucus. Positively charged sodium ions follow passively, increasing the total electrolyte concentration in the mucus, resulting in the movement of water out of the cell via osmosis.
In epithelial cells with motile cilia lining the bronchus and the oviduct, CFTR is located on the cell membrane but not on cilia. In contrast, ENaC (Epithelial sodium channel) is located along the entire length of the cilia.[26]
In sweat glands, defective CFTR results in reduced transport of sodium chloride and sodium thiocyanate[27] in the reabsorptive duct and therefore saltier sweat. This is the basis of a clinically important sweat test for cystic fibrosis often used diagnostically with genetic screening.[28]
Interactions
Cystic fibrosis transmembrane conductance regulator has been shown to interact with:
DNAJC5,[29]
GOPC,[30][31][31]
PDZK1,[31][32]
PRKCE,[33]
SLC4A8,[34]
SNAP23,[35]
SLC9A3R1,[19][34][36][37][38][39]
SLC9A3R2,[40] and
STX1A,[35][41]
It is inhibited by the anti-diarrhoea drug crofelemer.
Related conditions
Congenital bilateral absence of vas deferens: Males with congenital bilateral absence of the vas deferens most often have a mild mutation (a change that allows partial function of the gene) in one copy of the CFTR gene and a cystic fibrosis-causing mutation in the other copy of CFTR.
Cystic fibrosis: More than 1,800 mutations in the CFTR gene have been found[42] but the majority of these have not been associated with cystic fibrosis.[citation needed] Most of these mutations either substitute one amino acid (a building block of proteins) for another amino acid in the CFTR protein or delete a small amount of DNA in the CFTR gene. The most common mutation, called ΔF508, is a deletion (Δ) of one amino acid (phenylalanine) at position 508 in the CFTR protein. This altered protein never reaches the cell membrane because it is degraded shortly after it is made. All disease-causing mutations in the CFTR gene prevent the channel from functioning properly, leading to a blockage of the movement of salt and water into and out of cells. As a result of this blockage, cells that line the passageways of the lungs, pancreas, and other organs produce abnormally thick, sticky mucus. This mucus obstructs the airways and glands, causing the characteristic signs and symptoms of cystic fibrosis. In addition, only thin mucus can be removed by cilia; thick mucus cannot, so it traps bacteria that give rise to chronic infections.
Cholera: ADP-ribosylation caused by cholera toxin results in increased production of cyclic AMP which in turn opens the CFTR channel which leads to oversecretion of Cl−. Na+ and H2O follow Cl− into the small intestine, resulting in dehydration and loss of electrolytes.[43]
Drug target
CFTR has been a drug target in efforts to find treatments for related conditions. Ivacaftor (trade name Kalydeco, developed as VX-770) is a drug approved by the FDA in 2012 for people with cystic fibrosis who have specific CFTR mutations[44][45] Ivacaftor was developed by Vertex Pharmaceuticals in conjunction with the Cystic Fibrosis Foundation and is the first drug that treats the underlying cause rather than the symptoms of the disease.[46] Called "the most important new drug of 2012",[47] and "a wonder drug"[48] it is one of the most expensive drugs, costing over US$300,000 per year, which has led to criticism of Vertex for the high cost.
References
^ abc GRCh38: Ensembl release 89: ENSG00000001626 - Ensembl, May 2017
^ abc GRCm38: Ensembl release 89: ENSMUSG00000041301 - Ensembl, May 2017
^ "Human PubMed Reference:"..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ "Mouse PubMed Reference:".
^ ab Gadsby DC, Vergani P, Csanády L (March 2006). "The ABC protein turned chloride channel whose failure causes cystic fibrosis". Nature. 440 (7083): 477–83. Bibcode:2006Natur.440..477G. doi:10.1038/nature04712. PMC 2720541. PMID 16554808.
^ ab Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N (September 1989). "Identification of the cystic fibrosis gene: chromosome walking and jumping". Science. 245 (4922): 1059–65. Bibcode:1989Sci...245.1059R. doi:10.1126/science.2772657. PMID 2772657.
^ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL (September 1989). "Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA". Science. 245 (4922): 1066–73. Bibcode:1989Sci...245.1066R. doi:10.1126/science.2475911. PMID 2475911.
^ Childers M, Eckel G, Himmel A, Caldwell J (2007). "A new model of cystic fibrosis pathology: lack of transport of glutathione and its thiocyanate conjugates". Medical Hypotheses. 68 (1): 101–12. doi:10.1016/j.mehy.2006.06.020. PMID 16934416.
^ Marcorelles P, Gillet D, Friocourt G, Ledé F, Samaison L, Huguen G, Ferec C (March 2012). "Cystic fibrosis transmembrane conductance regulator protein expression in the male excretory duct system during development". Human Pathology. 43 (3): 390–7. doi:10.1016/j.humpath.2011.04.031. PMID 21840567.
^ ab "OrthoMaM phylogenetic marker: CFTR coding sequence".
^ Davies, R; Conroy, S-J; Davies, WL; Potter, IC; Rrezise, Ann EO (19–23 June 2005). "Evolution and Regulation of the Cystic Fibrosis Gene" (conference paper). Molecular Biology and Evolution (MBE05) Conference. Retrieved 28 July 2014.
^ Prasad AB, Allard MW, Green ED (September 2008). "Confirming the phylogeny of mammals by use of large comparative sequence data sets". Molecular Biology and Evolution. 25 (9): 1795–808. doi:10.1093/molbev/msn104. PMC 2515873. PMID 18453548.
^ "The Clinical and Functional TRanslation of CFTR (CFTR2): CFTR2 Variant List History". US CF Foundation, Johns Hopkins University, Cystic Fibrosis Centre at the Hospital for Sick Children in Toronto. Retrieved 2 August 2017.
^ Rowe SM, Miller S, Sorscher EJ (May 2005). "Cystic fibrosis". The New England Journal of Medicine. 352 (19): 1992–2001. doi:10.1056/NEJMra043184. PMID 15888700.
^ Kavic SM, Frehm EJ, Segal AS (1999). "Case studies in cholera: lessons in medical history and science". The Yale Journal of Biology and Medicine. 72 (6): 393–408. PMC 2579035. PMID 11138935.
^ Araújo FG, Novaes FC, Santos NP, Martins VC, Souza SM, Santos SE, Ribeiro-dos-Santos AK (January 2005). "Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil". Brazilian Journal of Medical and Biological Research = Revista Brasileira De Pesquisas Medicas E Biologicas. 38 (1): 11–5. doi:10.1590/S0100-879X2005000100003. PMID 15665983.
^ Cystic Fibrosis Mutation Database. "Genomic DNA sequence".
^ Sheppard DN, Welsh MJ (January 1999). "Structure and function of the CFTR chloride channel". Physiological Reviews. 79 (1 Suppl): S23–45. doi:10.1152/physrev.1999.79.1.S23. PMID 9922375.
^ ab Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL (July 1998). "An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton". The Journal of Biological Chemistry. 273 (31): 19797–801. doi:10.1074/jbc.273.31.19797. PMID 9677412.
^ Tsui LC, Dorfman R (February 2013). "The cystic fibrosis gene: a molecular genetic perspective". Cold Spring Harbor Perspectives in Medicine. 3 (2): a009472. doi:10.1101/cshperspect.a009472. PMC 3552342. PMID 23378595.
^ Tsai MF (October 2012). "CFTR: an ion channel with a transporter-type energy-coupling mechanism". The Journal of General Physiology. 140 (4): 343–5. doi:10.1085/jgp.201210882. PMC 3457686. PMID 22966013.
^ Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, Rogel N, Burgin G, Tsankov AM, Waghray A, Slyper M, Waldman J, Nguyen L, Dionne D, Rozenblatt-Rosen O, Tata PR, Mou H, Shivaraju M, Bihler H, Mense M, Tearney GJ, Rowe SM, Engelhardt JF, Regev A, Rajagopal J (August 2018). "A revised airway epithelial hierarchy includes CFTR-expressing ionocytes". Nature. 560 (7718): 319–324. doi:10.1038/s41586-018-0393-7. PMID 30069044.
^ Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, Klein AM, Jaffe AB (August 2018). "A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte". Nature. 560 (7718): 377–381. doi:10.1038/s41586-018-0394-6. PMID 30069046.
^ ab Hanukoglu I, Boggula VR, Vaknine H, Sharma S, Kleyman T, Hanukoglu A (June 2017). "Expression of epithelial sodium channel (ENaC) and CFTR in the human epidermis and epidermal appendages". Histochemistry and Cell Biology. 147 (6): 733–748. doi:10.1007/s00418-016-1535-3. PMID 28130590.
^ Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Nauseef WM, Dupuy C, Bánfi B (January 2007). "A novel host defense system of airways is defective in cystic fibrosis". American Journal of Respiratory and Critical Care Medicine. 175 (2): 174–83. doi:10.1164/rccm.200607-1029OC. PMC 2720149. PMID 17082494.
^ Enuka Y, Hanukoglu I, Edelheit O, Vaknine H, Hanukoglu A (March 2012). "Epithelial sodium channels (ENaC) are uniformly distributed on motile cilia in the oviduct and the respiratory airways". Histochemistry and Cell Biology. 137 (3): 339–53. doi:10.1007/s00418-011-0904-1. PMID 22207244.
^ Xu Y, Szép S, Lu Z (December 2009). "The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases". Proceedings of the National Academy of Sciences of the United States of America. 106 (48): 20515–9. Bibcode:2009PNAS..10620515X. doi:10.1073/pnas.0911412106. PMC 2777967. PMID 19918082.
^ Yonei Y, Tanaka M, Ozawa Y, Miyazaki K, Tsukada N, Inada S, Inagaki Y, Miyamoto K, Suzuki O, Okawa H (April 1992). "Primary hepatocellular carcinoma with severe hypoglycemia: involvement of insulin-like growth factors". Liver. 12 (2): 90–3. doi:10.1111/j.1600-0676.1992.tb00563.x. PMID 1320177.
^ Zhang H, Peters KW, Sun F, Marino CR, Lang J, Burgoyne RD, Frizzell RA (August 2002). "Cysteine string protein interacts with and modulates the maturation of the cystic fibrosis transmembrane conductance regulator". The Journal of Biological Chemistry. 277 (32): 28948–58. doi:10.1074/jbc.M111706200. PMID 12039948.
^ Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, Cutting GR, Li M, Stanton BA, Guggino WB (February 2002). "A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression". The Journal of Biological Chemistry. 277 (5): 3520–9. doi:10.1074/jbc.M110177200. PMID 11707463.
^ abc Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR (February 2003). "The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins". The Journal of Biological Chemistry. 278 (8): 6440–9. doi:10.1074/jbc.M211050200. PMID 12471024.
^ Wang S, Yue H, Derin RB, Guggino WB, Li M (September 2000). "Accessory protein facilitated CFTR-CFTR interaction, a molecular mechanism to potentiate the chloride channel activity". Cell. 103 (1): 169–79. doi:10.1016/S0092-8674(00)00096-9. PMID 11051556.
^ Liedtke CM, Yun CH, Kyle N, Wang D (June 2002). "Protein kinase C epsilon-dependent regulation of cystic fibrosis transmembrane regulator involves binding to a receptor for activated C kinase (RACK1) and RACK1 binding to Na+/H+ exchange regulatory factor". The Journal of Biological Chemistry. 277 (25): 22925–33. doi:10.1074/jbc.M201917200. PMID 11956211.
^ ab Park M, Ko SB, Choi JY, Muallem G, Thomas PJ, Pushkin A, Lee MS, Kim JY, Lee MG, Muallem S, Kurtz I (December 2002). "The cystic fibrosis transmembrane conductance regulator interacts with and regulates the activity of the HCO3- salvage transporter human Na+-HCO3- cotransport isoform 3". The Journal of Biological Chemistry. 277 (52): 50503–9. doi:10.1074/jbc.M201862200. PMID 12403779.
^ ab Cormet-Boyaka E, Di A, Chang SY, Naren AP, Tousson A, Nelson DJ, Kirk KL (September 2002). "CFTR chloride channels are regulated by a SNAP-23/syntaxin 1A complex". Proceedings of the National Academy of Sciences of the United States of America. 99 (19): 12477–82. Bibcode:2002PNAS...9912477C. doi:10.1073/pnas.192203899. PMC 129470. PMID 12209004.
^ Hegedüs T, Sessler T, Scott R, Thelin W, Bakos E, Váradi A, Szabó K, Homolya L, Milgram SL, Sarkadi B (March 2003). "C-terminal phosphorylation of MRP2 modulates its interaction with PDZ proteins". Biochemical and Biophysical Research Communications. 302 (3): 454–61. doi:10.1016/S0006-291X(03)00196-7. PMID 12615054.
^ Wang S, Raab RW, Schatz PJ, Guggino WB, Li M (May 1998). "Peptide binding consensus of the NHE-RF-PDZ1 domain matches the C-terminal sequence of cystic fibrosis transmembrane conductance regulator (CFTR)". FEBS Letters. 427 (1): 103–8. doi:10.1016/S0014-5793(98)00402-5. PMID 9613608.
^ Moyer BD, Duhaime M, Shaw C, Denton J, Reynolds D, Karlson KH, Pfeiffer J, Wang S, Mickle JE, Milewski M, Cutting GR, Guggino WB, Li M, Stanton BA (September 2000). "The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane". The Journal of Biological Chemistry. 275 (35): 27069–74. doi:10.1074/jbc.M004951200. PMID 10852925.
^ Hall RA, Ostedgaard LS, Premont RT, Blitzer JT, Rahman N, Welsh MJ, Lefkowitz RJ (July 1998). "A C-terminal motif found in the beta2-adrenergic receptor, P2Y1 receptor and cystic fibrosis transmembrane conductance regulator determines binding to the Na+/H+ exchanger regulatory factor family of PDZ proteins". Proceedings of the National Academy of Sciences of the United States of America. 95 (15): 8496–501. Bibcode:1998PNAS...95.8496H. doi:10.1073/pnas.95.15.8496. PMC 21104. PMID 9671706.
^ Sun F, Hug MJ, Lewarchik CM, Yun CH, Bradbury NA, Frizzell RA (September 2000). "E3KARP mediates the association of ezrin and protein kinase A with the cystic fibrosis transmembrane conductance regulator in airway cells". The Journal of Biological Chemistry. 275 (38): 29539–46. doi:10.1074/jbc.M004961200. PMID 10893422.
^ Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, Kirk KL (November 1997). "Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms". Nature. 390 (6657): 302–5. Bibcode:1997Natur.390..302N. doi:10.1038/36882. PMID 9384384.
^ Egan ME (March 2016). "Genetics of Cystic Fibrosis: Clinical Implications". Clinics in Chest Medicine. 37 (1): 9–16. doi:10.1016/j.ccm.2015.11.002. PMID 26857764.
^ Thiagarajah JR, Verkman AS (September 2012). "CFTR inhibitors for treating diarrheal disease". Clinical Pharmacology and Therapeutics. 92 (3): 287–90. doi:10.1038/clpt.2012.114. PMC 3643514. PMID 22850599.
^ Jones AM, Helm JM (October 2009). "Emerging treatments in cystic fibrosis". Drugs. 69 (14): 1903–10. doi:10.2165/11318500-000000000-00000. PMID 19747007.
^ McPhail GL, Clancy JP (April 2013). "Ivacaftor: the first therapy acting on the primary cause of cystic fibrosis". Drugs of Today. 49 (4): 253–60. doi:10.1358/dot.2013.49.4.1940984. PMID 23616952.
^ "Phase 3 Study of VX-770 Shows Marked Improvement in Lung Function Among People with Cystic Fibrosis with G551D Mutation". Press Release. Cystic Fibrosis Foundation. 2011-02-23.
^ Herper M (27 December 2012). "The Most Important New Drug Of 2012". Forbes.
^ Nocera J (18 July 2014). "The $300,000 Drug". New York Times.
Further reading
.mw-parser-output .refbegin{font-size:90%;margin-bottom:0.5em}.mw-parser-output .refbegin-hanging-indents>ul{list-style-type:none;margin-left:0}.mw-parser-output .refbegin-hanging-indents>ul>li,.mw-parser-output .refbegin-hanging-indents>dl>dd{margin-left:0;padding-left:3.2em;text-indent:-3.2em;list-style:none}.mw-parser-output .refbegin-100{font-size:100%}
Kulczycki LL, Kostuch M, Bellanti JA (January 2003). "A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations". American Journal of Medical Genetics. Part A. 116A (3): 262–7. doi:10.1002/ajmg.a.10886. PMID 12503104.
Vankeerberghen A, Cuppens H, Cassiman JJ (March 2002). "The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions". Journal of Cystic Fibrosis. 1 (1): 13–29. doi:10.1016/S1569-1993(01)00003-0. PMID 15463806.
Tsui LC (1992). "Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the Cystic Fibrosis Genetic Analysis Consortium". Human Mutation. 1 (3): 197–203. doi:10.1002/humu.1380010304. PMID 1284534.
McIntosh I, Cutting GR (July 1992). "Cystic fibrosis transmembrane conductance regulator and the etiology and pathogenesis of cystic fibrosis". FASEB Journal. 6 (10): 2775–82. PMID 1378801.
Drumm ML, Collins FS (1993). "Molecular biology of cystic fibrosis". Molecular Genetic Medicine. 3: 33–68. doi:10.1016/b978-0-12-462003-2.50006-7. PMID 7693108.
Kerem B, Kerem E (1996). "The molecular basis for disease variability in cystic fibrosis". European Journal of Human Genetics. 4 (2): 65–73. PMID 8744024.
Devidas S, Guggino WB (October 1997). "CFTR: domains, structure, and function". Journal of Bioenergetics and Biomembranes. 29 (5): 443–51. doi:10.1023/A:1022430906284. PMID 9511929.
Nagel G (December 1999). "Differential function of the two nucleotide binding domains on cystic fibrosis transmembrane conductance regulator". Biochimica et Biophysica Acta. 1461 (2): 263–74. doi:10.1016/S0005-2736(99)00162-5. PMID 10581360.
Boyle MP (2000). "Unique presentations and chronic complications in adult cystic fibrosis: do they teach us anything about CFTR?". Respiratory Research. 1 (3): 133–5. doi:10.1186/rr23. PMC 59552. PMID 11667976.
Greger R, Schreiber R, Mall M, Wissner A, Hopf A, Briel M, Bleich M, Warth R, Kunzelmann K (2001). "Cystic fibrosis and CFTR". Pflügers Archiv. 443 Suppl 1: S3–7. doi:10.1007/s004240100635. PMID 11845294.
Bradbury NA (2001). "cAMP signaling cascades and CFTR: is there more to learn?". Pflügers Archiv. 443 Suppl 1: S85–91. doi:10.1007/s004240100651. PMID 11845310.
Dahan D, Evagelidis A, Hanrahan JW, Hinkson DA, Jia Y, Luo J, Zhu T (2001). "Regulation of the CFTR channel by phosphorylation". Pflügers Archiv. 443 Suppl 1: S92–6. doi:10.1007/s004240100652. PMID 11845311.
Cohn JA, Noone PG, Jowell PS (September 2002). "Idiopathic pancreatitis related to CFTR: complex inheritance and identification of a modifier gene". Journal of Investigative Medicine. 50 (5): 247S–255S. doi:10.1136/jim-50-suppl5-01. PMID 12227654.
Schwartz M (February 2003). "[Cystic fibrosis transmembrane conductance regulator (CFTR) gene: mutations and clinical phenotypes]". Ugeskrift for Laeger. 165 (9): 912–6. PMID 12661515.
Wong LJ, Alper OM, Wang BT, Lee MH, Lo SY (July 2003). "Two novel null mutations in a Taiwanese cystic fibrosis patient and a survey of East Asian CFTR mutations". American Journal of Medical Genetics. Part A. 120A (2): 296–8. doi:10.1002/ajmg.a.20039. PMID 12833420.
Cuppens H, Cassiman JJ (October 2004). "CFTR mutations and polymorphisms in male infertility". International Journal of Andrology. 27 (5): 251–6. doi:10.1111/j.1365-2605.2004.00485.x. PMID 15379964.
Cohn JA, Mitchell RM, Jowell PS (March 2005). "The impact of cystic fibrosis and PSTI/SPINK1 gene mutations on susceptibility to chronic pancreatitis". Clinics in Laboratory Medicine. 25 (1): 79–100. doi:10.1016/j.cll.2004.12.007. PMID 15749233.
Southern KW, Peckham D (2004). "Establishing a diagnosis of cystic fibrosis". Chronic Respiratory Disease. 1 (4): 205–10. doi:10.1191/1479972304cd044rs. PMID 16281647.
Kandula L, Whitcomb DC, Lowe ME (June 2006). "Genetic issues in pediatric pancreatitis". Current Gastroenterology Reports. 8 (3): 248–53. doi:10.1007/s11894-006-0083-8. PMID 16764792.
Marcet B, Boeynaems JM (December 2006). "Relationships between cystic fibrosis transmembrane conductance regulator, extracellular nucleotides and cystic fibrosis". Pharmacology & Therapeutics. 112 (3): 719–32. doi:10.1016/j.pharmthera.2006.05.010. PMID 16828872.
Wilschanski M, Durie PR (August 2007). "Patterns of GI disease in adulthood associated with mutations in the CFTR gene". Gut. 56 (8): 1153–63. doi:10.1136/gut.2004.062786. PMC 1955522. PMID 17446304.
External links
- GeneReviews/NCBI/NIH/UW entry on CFTR-Related Disorders - Cystic Fibrosis (CF, Mucoviscidosis) and Congenital Absence of the Vas Deferens (CAVD)
- The Cystic Fibrosis Transmembrane Conductance Regulator Protein
- The Human Gene Mutation Database - CFTR Records
- Cystic Fibrosis Mutation Database
- Oak Ridge National Laboratory CFTR Information
- CFTR at OMIM (National Center for Biotechnology Information)
PDB gallery | |
|---|---|
|